RNA-Seq components, airway dataset, using cpm+vooma
airway_vooma.RmdLet’s look at the airway dataset as an example of a typical small-scale RNA-Seq experiment. In this experiment, four Airway Smooth Muscle (ASM) cell lines are treated with the asthma medication dexamethasone.
library(weitrix)
library(SummarizedExperiment)
library(EnsDb.Hsapiens.v86)
library(edgeR)
library(limma)
library(reshape2)
library(tidyverse)
library(airway)
set.seed(1234)data("airway")
airway## class: RangedSummarizedExperiment
## dim: 64102 8
## metadata(1): ''
## assays(1): counts
## rownames(64102): ENSG00000000003 ENSG00000000005 ... LRG_98 LRG_99
## rowData names(0):
## colnames(8): SRR1039508 SRR1039509 ... SRR1039520 SRR1039521
## colData names(9): SampleName cell ... Sample BioSampleInitial processing
Initial steps are the same as for a differential expression analysis.
counts <- assay(airway,"counts")
# Keep genes with an average of at least 1 read per sample
good <- rowMeans(counts) >= 1
table(good)## good
## FALSE TRUE
## 40931 23171airway_cpm <-
DGEList(counts[good,]) %>%
calcNormFactors() %>%
cpm(log=TRUE, prior.count=1)
design <- model.matrix(~ dex + cell, data=colData(airway))
airway_elist <- vooma(airway_cpm, design=design, plot=TRUE)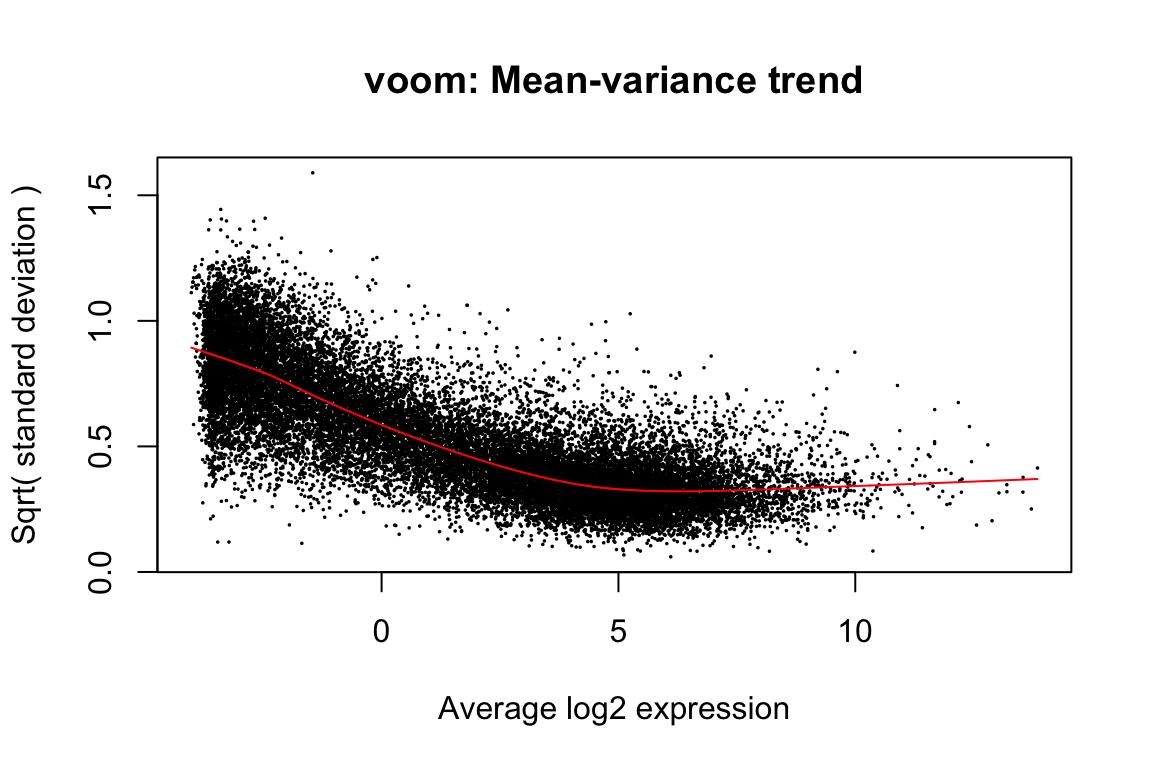
Some of these steps are optional and have marginal value:
voomaprovides precision weights. One could instead choose aprior.countforcpmto produce a flat (or at least non-decreasing) mean-variance trend-line.Using a design matrix with
voomamay not provide much benefit, and if you want an analysis completely blinded to the experimental design it might be better to omit this.
There are many options here, so adjust to your taste!
Conversion to weitrix
airway_weitrix <- as_weitrix(airway_elist)
# Include row and column information
colData(airway_weitrix) <- colData(airway)
rowData(airway_weitrix) <-
mcols(genes(EnsDb.Hsapiens.v86))[rownames(airway_weitrix),c("gene_name","gene_biotype")]
airway_weitrix## class: SummarizedExperiment
## dim: 23171 8
## metadata(1): weitrix
## assays(2): x weights
## rownames(23171): ENSG00000000003 ENSG00000000419 ... ENSG00000273487
## ENSG00000273488
## rowData names(2): gene_name gene_biotype
## colnames(8): SRR1039508 SRR1039509 ... SRR1039520 SRR1039521
## colData names(9): SampleName cell ... Sample BioSampleExploration
Find components of variation
This will find various numbers of components, from 1 to 6. In each case, the components discovered have varimax rotation applied to their gene loadings to aid interpretability. The result is a list of Components objects.
comp_seq <- weitrix_components_seq(airway_weitrix, p=6)
comp_seq## [[1]]
## Components are: (Intercept), C1
## $row : 23171 x 2 matrix
## $col : 8 x 2 matrix
## $R2 : 0.3990789
##
## [[2]]
## Components are: (Intercept), C1, C2
## $row : 23171 x 3 matrix
## $col : 8 x 3 matrix
## $R2 : 0.6229488
##
## [[3]]
## Components are: (Intercept), C1, C2, C3
## $row : 23171 x 4 matrix
## $col : 8 x 4 matrix
## $R2 : 0.790927
##
## [[4]]
## Components are: (Intercept), C1, C2, C3, C4
## $row : 23171 x 5 matrix
## $col : 8 x 5 matrix
## $R2 : 0.8948288
##
## [[5]]
## Components are: (Intercept), C1, C2, C3, C4, C5
## $row : 23171 x 6 matrix
## $col : 8 x 6 matrix
## $R2 : 0.9388779
##
## [[6]]
## Components are: (Intercept), C1, C2, C3, C4, C5, C6
## $row : 23171 x 7 matrix
## $col : 8 x 7 matrix
## $R2 : 0.972438rand_weitrix <- weitrix_randomize(airway_weitrix)
rand_comp <- weitrix_components(rand_weitrix, p=1)
components_seq_screeplot(comp_seq, rand_comp)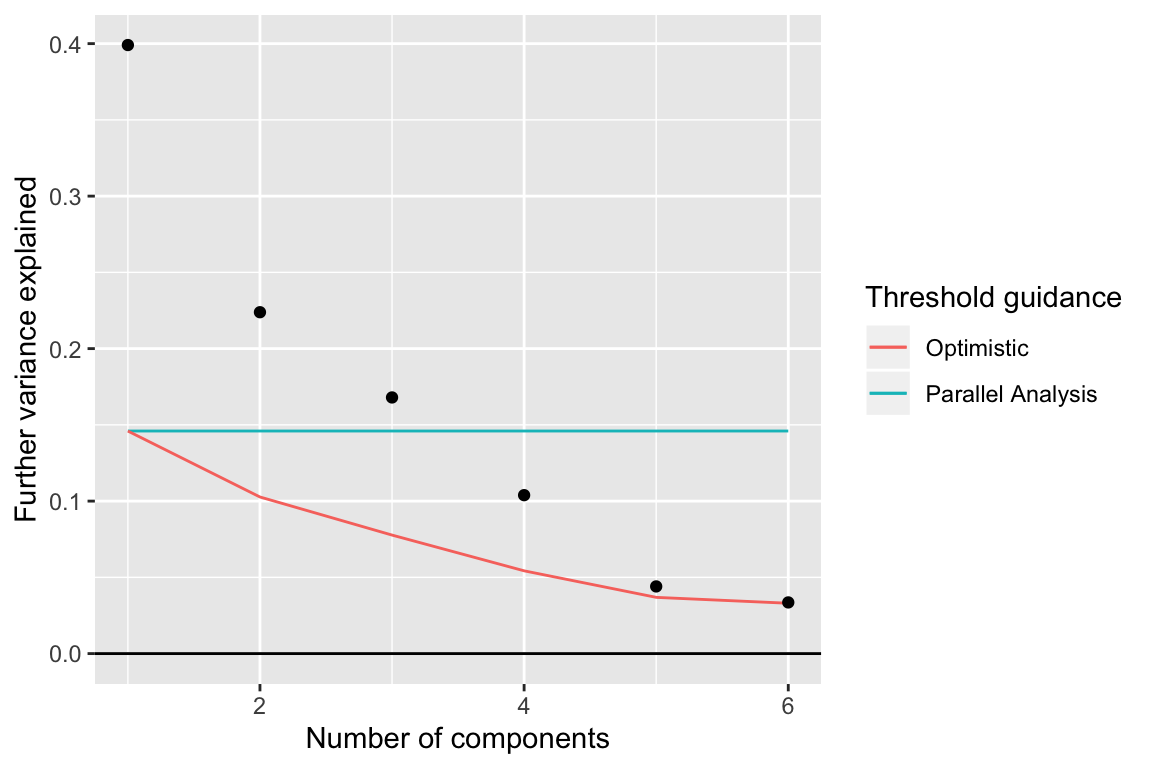
Examining components
Up to 4 components may be justified.
comp <- comp_seq[[4]]
comp$col[,-1] %>% melt(varnames=c("Run","component")) %>%
left_join(as.data.frame(colData(airway)), by="Run") %>%
ggplot(aes(y=cell, x=value, color=dex)) +
geom_vline(xintercept=0) +
geom_point(alpha=0.5, size=3) +
facet_grid(~ component) +
labs(title="Sample scores for each component", x="Sample score", y="Cell line", color="Treatment")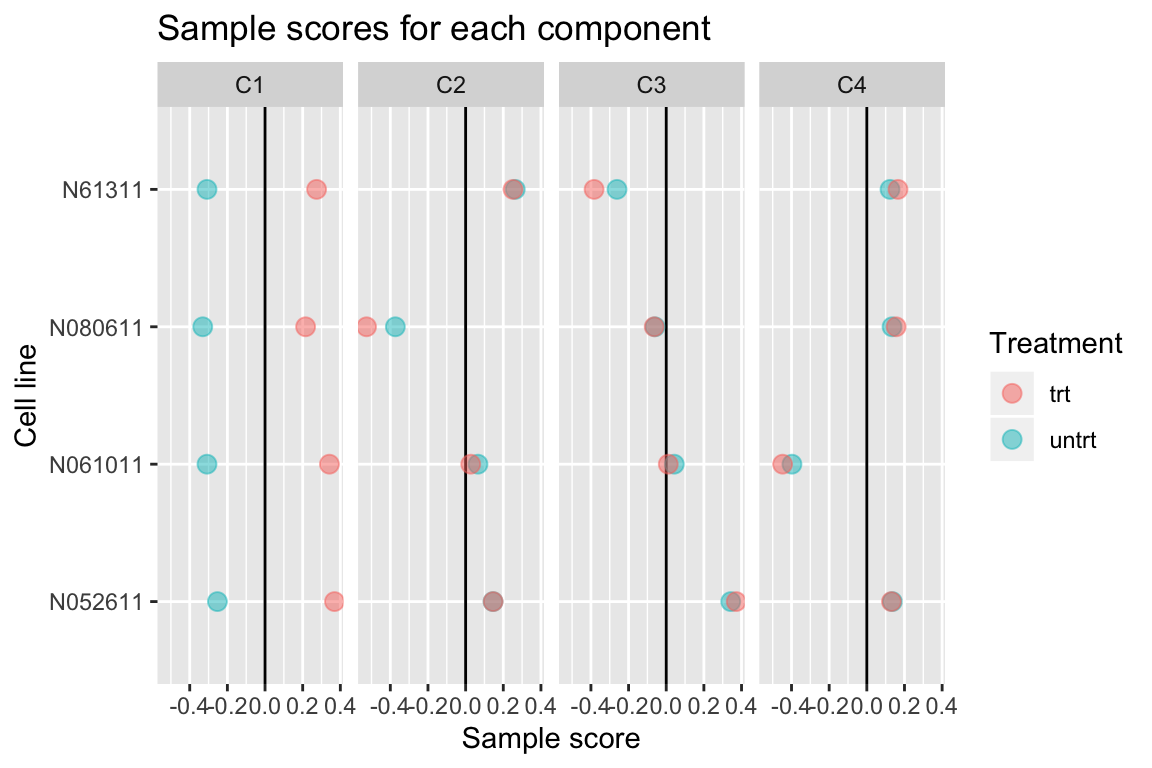
comp$row[,-1] %>% melt(varnames=c("name","component")) %>%
ggplot(aes(x=comp$row[name,"(Intercept)"], y=value)) +
geom_point(cex=0.5, alpha=0.5) +
facet_wrap(~ component) +
labs(title="Gene loadings for each component vs average log2 RPM", x="average log2 RPM", y="gene loading")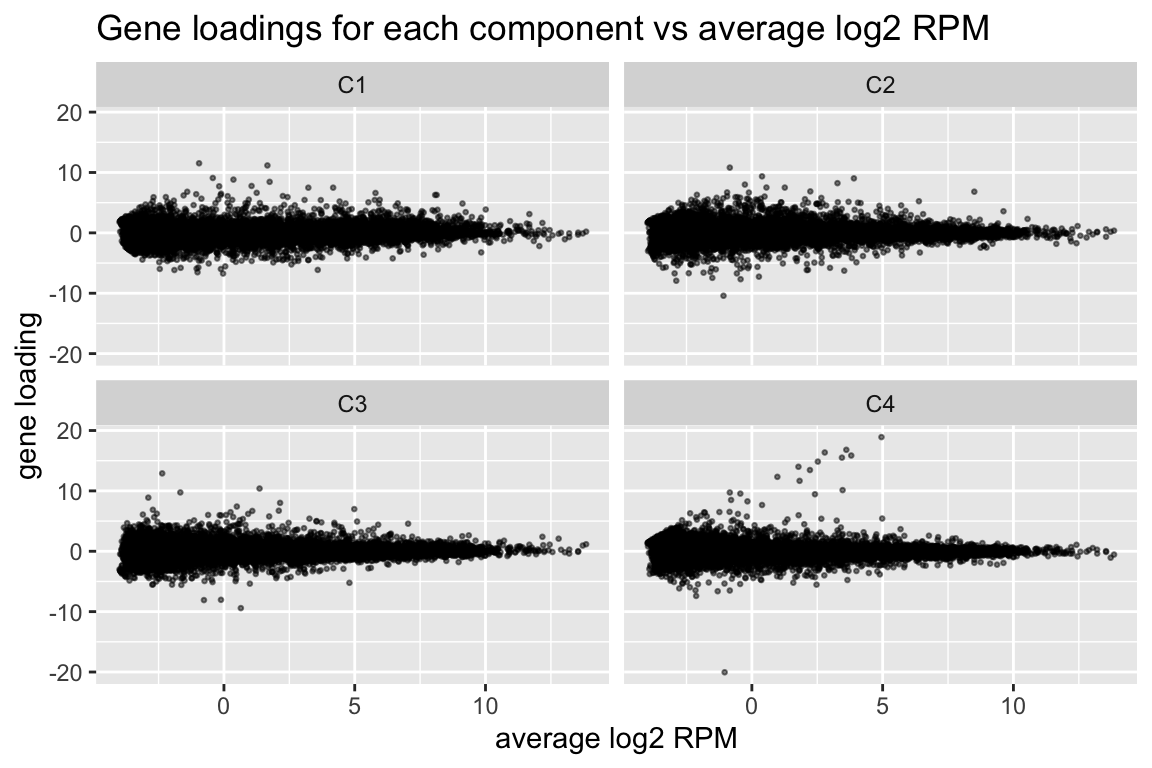
Without varimax rotation, components may be harder to interpret
If varimax rotation isn’t used, weitrix_components and weitrix_components_seq will produce a Principal Components Analysis, with components ordered from most to least variance explained.
Without varimax rotation the treatment effect is still mostly in the first component, but has also leaked a small amount into the other components.
comp_nonvarimax <- weitrix_components(airway_weitrix, p=4, use_varimax=FALSE)
comp_nonvarimax$col[,-1] %>% melt(varnames=c("Run","component")) %>%
left_join(as.data.frame(colData(airway)), by="Run") %>%
ggplot(aes(y=cell, x=value, color=dex)) +
geom_vline(xintercept=0) +
geom_point(alpha=0.5, size=3) +
facet_grid(~ component) +
labs(title="Sample scores for each component, no varimax rotation", x="Sample score", y="Cell line", color="Treatment")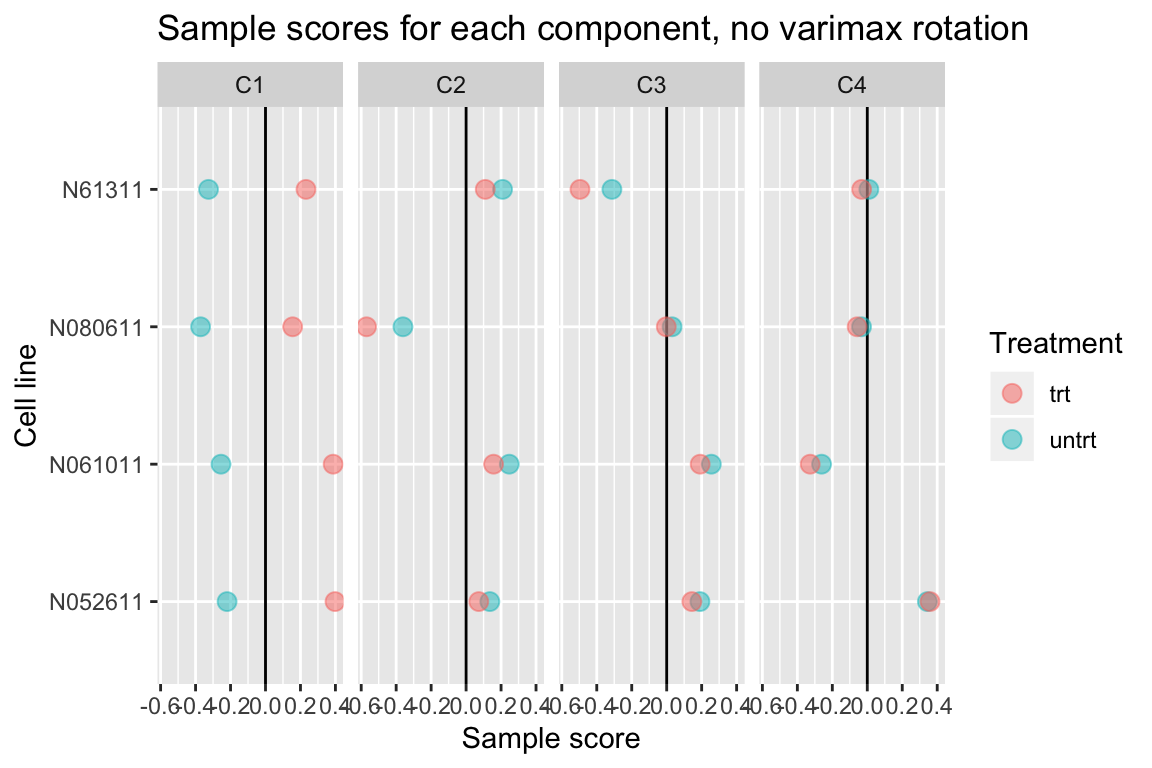
col can potentially be used as a design matrix with limma
If you are willing to place your trust in sparsity of effects, this may be a somewhat magical way of removing unwanted variation.
If you’re not sure of the experimental design, for example the exact timing of a time series or how evenly a drug treatment was applied, the extracted component might actually be more accurate.
Note that this ignores uncertainty about the col matrix itself.
This may be useful for hypothesis generation – finding some potentially interesting genes, while discounting noisy or lowly expressed genes – but don’t use it as proof of significance.
airway_elist <- weitrix_elist(airway_weitrix)
fit <-
lmFit(airway_elist, comp$col) %>%
eBayes()
fit$df.prior## [1] 8.595638## [1] 1.076146topTable(fit, "C1")## gene_name gene_biotype logFC AveExpr t
## ENSG00000101347 SAMHD1 protein_coding 6.283933 8.134279 48.58913
## ENSG00000189221 MAOA protein_coding 5.482452 5.950311 36.06758
## ENSG00000178695 KCTD12 protein_coding -4.227635 6.450973 -32.96743
## ENSG00000139132 FGD4 protein_coding 3.652991 5.415156 31.41780
## ENSG00000120129 DUSP1 protein_coding 4.895673 6.643767 31.01784
## ENSG00000179094 PER1 protein_coding 5.204844 4.421867 30.98882
## ENSG00000124766 SOX4 protein_coding -4.084065 5.426691 -29.55701
## ENSG00000162692 VCAM1 protein_coding -6.134506 3.575005 -31.34264
## ENSG00000134243 SORT1 protein_coding 3.596138 7.568421 27.48157
## ENSG00000163884 KLF15 protein_coding 7.483922 3.248049 31.70319
## P.Value adj.P.Val B
## ENSG00000101347 9.185497e-15 2.128371e-10 23.17495
## ENSG00000189221 2.848140e-13 3.299712e-09 20.50857
## ENSG00000178695 8.000298e-13 4.714209e-09 19.72905
## ENSG00000139132 1.390192e-12 4.714209e-09 19.19450
## ENSG00000120129 1.610249e-12 4.714209e-09 19.10150
## ENSG00000179094 1.627624e-12 4.714209e-09 18.69549
## ENSG00000124766 2.799112e-12 7.022876e-09 18.54653
## ENSG00000162692 1.428917e-12 4.714209e-09 18.11379
## ENSG00000134243 6.439868e-12 1.243485e-08 17.84781
## ENSG00000163884 1.253204e-12 4.714209e-09 17.75410all_top <- topTable(fit, "C1", n=Inf, sort.by="none")
plotMD(fit, "C1", status=all_top$adj.P.Val <= 0.01)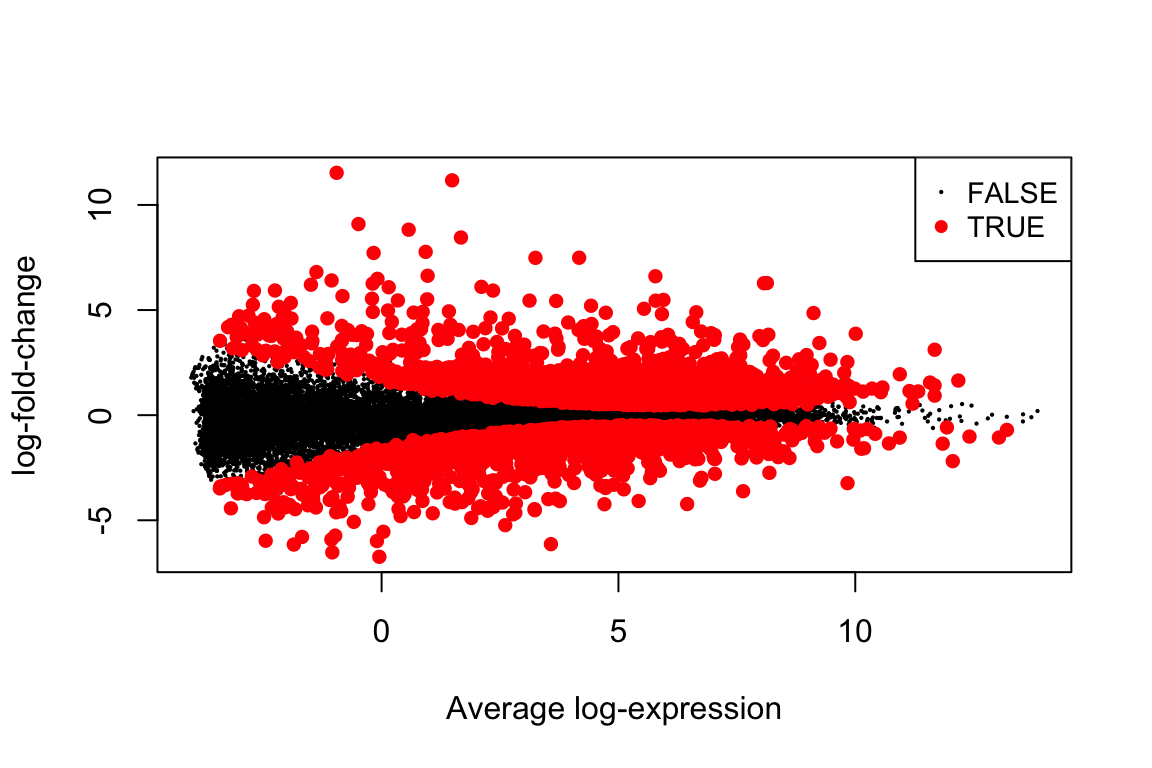
You might also consider using my topconfects package. This will find the largest confident effect sizes, while still correcting for multiple testing.
library(topconfects)
limma_confects(fit, "C1")## $table
## rank index confect effect AveExpr name gene_name
## 1 12120 6.665 11.528546 -0.9495193 ENSG00000179593 ALOX15B
## 2 3272 6.608 11.169008 1.4890296 ENSG00000109906 ZBTB16
## 3 11016 5.832 8.822691 0.5713077 ENSG00000171819 ANGPTL7
## 4 9499 5.832 7.483922 3.2480486 ENSG00000163884 KLF15
## 5 8175 5.744 7.488661 4.1705059 ENSG00000152583 SPARCL1
## 6 2230 5.416 6.283933 8.1342789 ENSG00000101347 SAMHD1
## 7 5156 5.375 8.450418 1.6752492 ENSG00000127954 STEAP4
## 8 10420 5.189 7.767901 0.9298793 ENSG00000168309 FAM107A
## 9 19752 4.935 9.090523 -0.4883206 ENSG00000250978 RP11-357D18.1
## 10 9220 -4.892 -6.134506 3.5750054 ENSG00000162692 VCAM1
## gene_biotype
## protein_coding
## protein_coding
## protein_coding
## protein_coding
## protein_coding
## protein_coding
## protein_coding
## protein_coding
## processed_transcript
## protein_coding
## ...
## 5534 of 23171 non-zero log2 fold change at FDR 0.05
## Prior df 8.6